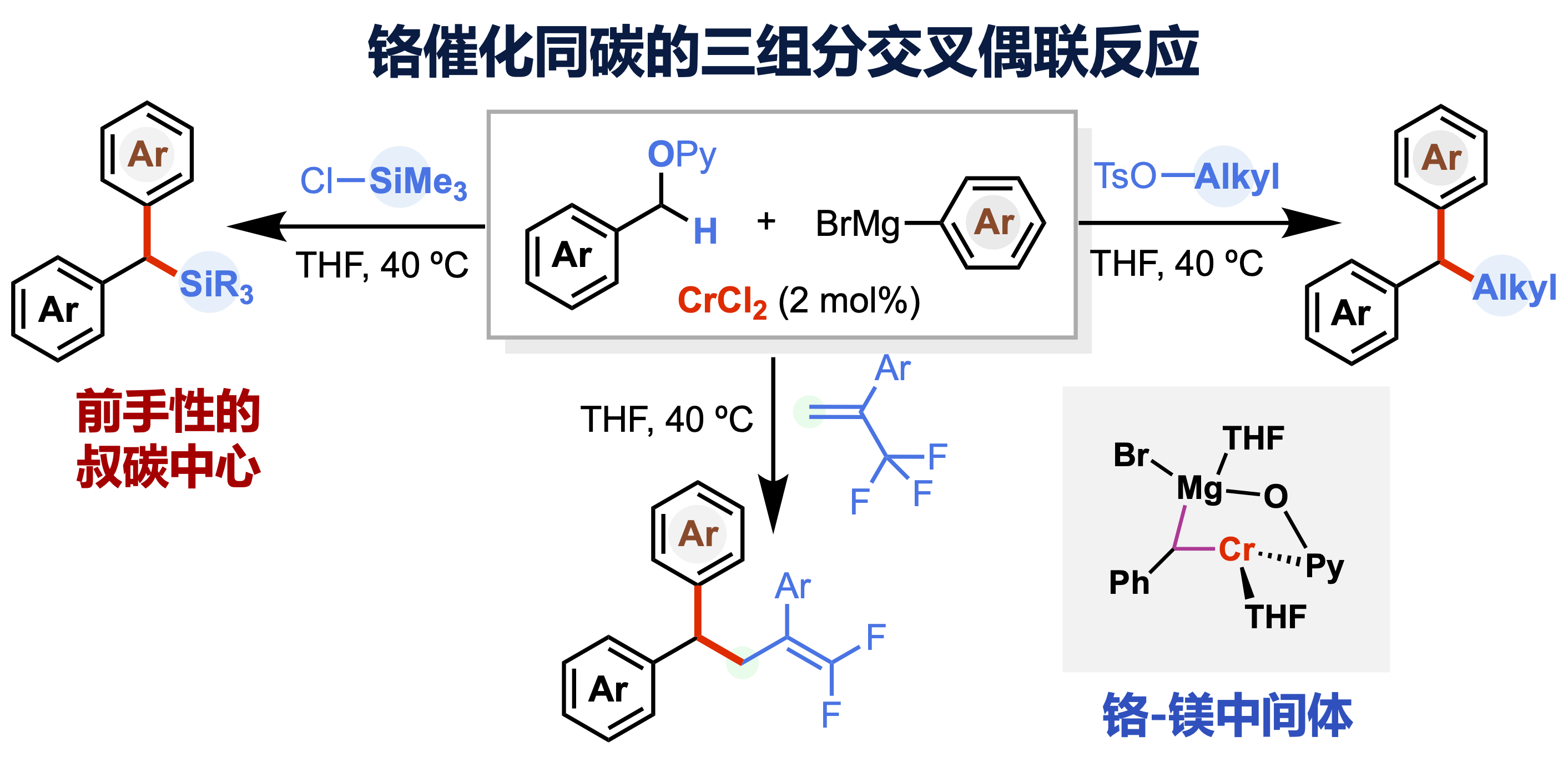
有机合成的核心任务是高效、高选择性地构筑重要功能化合物。传统的交叉偶联常涉及两组分反应一步生成一根化学键(one bond formation)。对应地,多组分交叉偶联可一步生成多根化学键,具有高效、快速及不用分离中间体的特点,但因其副反应较多,面临化学选择性挑战。值得注意的是,通过断裂同碳位置上两根不同的化学键,将其与亲电和亲核试剂同时偶联构筑前手性的叔碳化合物,仍缺乏有效的合成手段。由于这类前手性叔碳中心广泛于众多药物分子的结构单元中,发展策略来实现三组分底物在同碳位置的交叉偶联、一步高效构筑叔碳化合物具有重要研究意义。
曾小明课题组结合前期发展的底物导向活化策略(J. Am. Chem. Soc. 2015, 137, 14367;J. Am. Chem. Soc. 2017, 139, 15182),最近以廉价的二氯化铬为催化剂前体,开发出苄基醚与格式试剂及另一种亲电试剂如氯硅烷、烷基酯或烯基三氟甲基之间在同碳位置的三组分交叉偶联新反应,化学选择性地构筑了取代多样的叔碳化合物。研究发现仅铬金属盐在该反应中表现出催化活性,筛选的众多金属盐如氯化钯、氯化钌、氯化镍等都不能促进反应的进行,在一定程度上体现出铬金属较独特的催化活性。机理研究表明原位生成的低价铬首先断裂惰性的碳-氧键,在格式试剂作为强碱攫取烷基铬的α-氢后,形成异核的铬-镁双金属中间体,进一步与格式试剂及氯硅烷反应在同碳位置生成两根化学键。该铬催化的三组分偶联策略已被应用于合成普罗扎平、芬哌丙烷、地索普明及芬地林等市售药物及多种具有抗癌活性化合物,展现出一定的药物化学应用前景。
该研究以"Chemoselective chromium-catalysed cross-coupling enables three-component tertiary alkane synthesis"发表于《Nature Synthesis》(文章链接:https://www.nature.com/articles/s44160-023-00364-w)。四川大学为第一单位,化学学院曾小明教授、罗美明教授、北京化学所陈辉研究员为本文通讯作者,文章第一作者是四川大学博士研究生凡飞,北化所陈辉研究员提供理论计算合作研究。特别感谢国家自然科学基金委及科技部对本项目研究提供的经费支持。
